Proteolysis targeting chimera (PROTAC) is a novel drug development strategy that has brought a paradigm shift as a therapeutic modality. This field has evolved greatly in recent years in drug discovery, riding a wave of new tools, novel algorithms, and growing investment to more widespread studies. Numerous biopharma organizations seek to take advantage of targeted protein degradation’s (TPD) advantages over small molecule inhibition, including the ability to maintain greater control over proteins with non-enzymatic roles and to target the undruggable space.Aragen offers the expertise and experience as a drug discovery partner to enable organizations to successfully execute and drive TPD-based programs.
Proteolysis targeting chimeras (PROTACs) are heterobifunctional molecules comprised of three components: an E3ligase ligand or recruiting element, target protein of interest (POI) ligand and a chemical linker joining the two. Binding of the PROTAC molecule to the target protein and E3 ligase eventually induces the formation of a ternary complex, followed by ubiquitination of lysine residues in the target protein and subsequent degradation mediated by the 26S proteasomal machinery [i].
Degradation is a significantly different modality from small molecule inhibition as PROTACs have a catalytic mode of action, eventdriven pharmacology, and ability to target undruggable proteins with shallow pockets like Kirsten rat sarcoma virus (KRAS), nonenzymatic scaffolding proteins, multicomponent protein complexes like the BAF chromatin remodelling complex and transcription factors [ii].
Since the first report of PROTACs in 2001, multiple degraders based on the PROTACs have entered clinical development and have demonstrated good safety and target degradation [iii]. Arvinas candidates ARV-110 (prostate cancer) and ARV-471 (breast cancer) are in clinical Phase 2, and ARV-766 (prostate cancer) is in clinical Phase 1, along with other degraders from Kymera, Dialectic Therapeutics, BMS, Foghorn Therapeutics, and Nurix Therapeutics in Phase 1 trials [iv]. Major pharmaceutical companies have invested in TPD field and over 25 TPD-focused organizations have emerged over the last few years [v].
The majority of clinical candidates have leveraged cereblon (CRBN) as the E3 ligase partner. Interestingly, the von Hippel-Lindau (VHL)-based degrader for Bcl-xL (DT2216) demonstrated the potential to reduce the thrombocytopenia-based toxicity seen with Bcl-xL inhibitors. This has been attributed to low expression of VHL in platelets — hence, less platelet toxicity. Thus, PROTACs can have advantages over their POI inhibitor counterparts if they have tissue-restricted expression.
Multiple E3 ligases should be tested during PROTAC development, as not all E3 ligases can degrade protein targets. PROTACs have the potential to have a high pharmacodynamic potency and longer duration of action for targets with slower resynthesis. Due to their catalytic nature, sustained high drug exposure is not required to drive desired PD effect in absence of PK-PD correlation.
Considering that about 600 E3 ligases have been identified, only a handful have been explored using a TPD modality. Current investment and exploration in TPD are heavily focused on investigating the E3 ligase repertoire because discovery to date has mainly been driven by CRBN and VHL, whose warheads have narrow structure–activity relationships (SARs) [v].
This necessitates the discovery of novel warheads targeting other E3 ligases and expansion of the E3 ligase toolbox [vi]. Recently, new E3 ligases RNF4, RNF114, DCAF16, and Keap1 have been added to that toolbox, and ligands are being developed with chemo proteomics and DNA-encoded library (DEL) screen-based approaches [i].
Moreover, there exist numerous undruggable target proteins, which have no enzymatic role, but they still have strong clinical function (e.g., scaffolding or signalling complexes) and can be therapeutically beneficial if inhibited. In recent years, heterobifunctional PROTAC molecules have emerged as degradation inducers for undruggable molecules [vii] [viii].
To discover a PROTAC molecule for a novel (undruggable) target and a novel E3 ligase, in addition to understanding the target’s biology (which applies to any modality), an approach must be devised to determine whether the target is degradable. Recently, chemical genetic studies have been employed using HaloPROTACs with HaloTag7 fusion proteins [xi].
The complexity of developing an effective degrader requires the availability of a target ligand and E3 ligase ligand. When executing a novel approach, multiple techniques are necessary including artificial intelligence and machine learning to design degraders [x].
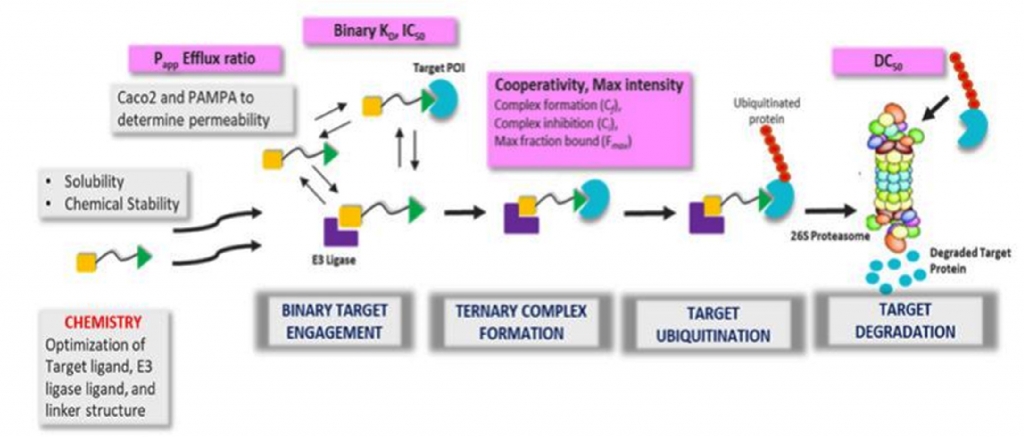
Effectively and efficiently understanding the degradation mechanism of action and how different E3 ligases can be leveraged requires having appropriate tools and technologies in place, as well as insight into which assays are to be developed within a screening funnel. We design in vitro assays to screen PROTAC-based degraders enabling characterization in cell-free and cell-based systems and deploy appropriate biophysical measurements to measure binding kinetics of binary complex and ternary complex formation. These biochemical assays measure the binary affinity, ternary complex formation, peak height, and cooperativity of PROTACs.
We have observed that PROTACs with weak binary interactions are potent degraders if they form a stable ternary complex. Several different technologies, including fluorescence polarization, amplified luminescent proximity homogeneous assays (ALPHA) technology, and time-resolved fluorescence energy transfer assays are used for routing screening [xi].
Additionally, biophysical techniques like surface plasmon resonance (SPR) [xii] are simultaneously used to measure the cooperativity (α = KDbinary /KDternary) factor, ternary binding affinity (Kd ) and kinetics (Kon , Koff and t1/2 ) of a ternary complex [xiii]. It has been demonstrated that formation of favourable and new protein-protein interactions significantly influences ternary complex stabilization.
In terms of cell-based degradation assays, the most straightforward method to assess degradation is based on reporter-tagged protein of interest (POI) expressing cell lines, or profiling degradation in selected cell lines where both POI and E3 ligase are expressed endogenously and PROTAC treatment will result in POI degradation. Cell lines expressing HiBit-tagged POI are used for routine screening and evaluation of Dmax and half maximal degradation concentrations or DC50 to rank order these molecules.
Dose response curves (DRCs) determining cellular DC50 PROTACs are evaluated at fixed time points or in a time course. As PROTAC molecules need to demonstrate endogenous target degradation in cell lines expressing the POI and E3 ligase, these data should be corroborated with experiments assessing ubiquitination of the target protein, involvement of proteasomal machinery by inclusion of proteasomal inhibitors to demonstrate rescue of degradation, and by using control cell lines lacking E3 ligase expression. If the protein is ubiquitinated, when is the protein’s next cycle of regeneration? A protein with a very fast turnaround time may be ineffective as a degrader since a protein expressed endogenously, with a longer turnaround time after degradation, provides a longer period of effective suppression of the target and downstream signalling.
At very high concentrations of PROTACs, hook effects may be observed in cells, consistent with a target protein-PROTAC or E3 ligase-PROTAC binary complex formation, which will abrogate degradation. Additional cellular assays are incorporated to monitor cellular target engagement, ternary complex formation and ubiquitination using proximity based NanoBRET technology from Promega
However, the identification of a degrader with the right combination of biological outcome and in vivo efficacy is a complex process, as PROTACs lie in chemical space beyond Lipinski’s Rule of Five due to high MW (bRo5), which suggests that achieving oral bioavailability will be challenging. Moreover, these bifunctional molecules are also associated with low solubility and permeability, high clearance, high non-specific binding, and chemical instability
Competition between PROTAC metabolites and PROTAC molecules for binding to POI and E3 ligase will also result in reduced degradation and efficacy. The polar nature of PROTAC warheads and high TPSA makes PROTACS less likely to be CNS penetrant. Since PROTACs have high lipophilicity, they are also expected to have low unbound fraction (fu). Hence, we emphasize screening of PROTAC-based molecules involving detailed characterization of physicochemical properties, solubility, permeability, and metabolic stability, as well as apply mitigation plans for optimization of ADME parameters.
Based on the scope of our collaborations, we offer flexible solutions to accommodate the varying queries and emphases of our partners. We provide the reagents, tools, and technologies to design an effective strategy to meet the project goals.
Our strategy is guided by the critical decision-making factors. Is plenty of prior knowledge already available about the target (e.g., it has been validated), or is its functionality a mystery? For example, if the target is novel or the downstream effects are not well-known, what is the differentiation in functional response if inhibited or degraded? We design appropriate functional and mechanistic assays to understand target biology and the outcome of target degradation.
As no project has an indefinite timeline, it is imperative that our collaboration produces clear, data-informed go/no-go decisions. Our experiences, successes, and (calculated) failures have sharpened our TPD focus, instilling confidence in our expertise to provide an endto-end solution for our collaborators. To learn more, contact the author and visit www.aragen.com.
Aragen is a global leader in providing drug discovery, drug development, and manufacturing solutions for life sciences firms.