Comprehensive profiling approaches, including genomics, transcriptomics, and proteomics, enable library-scale characterization of molecular alterations in response to various stimuli, such as drug treatment. Among these, quantitative proteomics plays a pivotal role in providing critical insights into protein dynamics, essential for understanding cellular processes, disease mechanisms, and therapeutic responses. These insights help reveal how cellular exposure to drugs and tool compounds induces changes in the proteomic signature, driven by both on-target and o-target e ects of test compounds.
However, in the post-genomic era, a key challenge is the complexity of technologies required for comprehensive proteome characterization. Proteomic analysis o ers a systematic approach to identifying and quantifying proteins at specific time points, allowing researchers to gain insights into cellular dynamics through protein profile comparisons, protein–protein interaction analysis, and organelle-specific proteomics. This global profiling helps identify protein markers associated with specific cell populations or drug-induced effects.
To address the inherent complexities of proteome analysis, stable isotopic labeling techniques like Tandem Mass Tags (TMT) have revolutionized the field of quantitative proteomics. This article focuses on TMT, a robust labeling technique that enables multiplexed analysis of proteomic samples and allows for high-throughput quantification without compromising sensitivity or accuracy. TMT labeling streamlines workflows while ensuring reproducibility and depth of analysis, making it an invaluable method for biomarker discovery, drug development, and personalized medicine. TMT enables the increasing demand for insights into cellular phenotypes, post-translational modifications, and protein function, which are vital for understanding disease mechanisms and drug targeting.
Proteomic analysis involves a series of steps, including the identification, separation, purification, sequencing, and characterization of proteins. Each of these steps relies on advanced techniques and bioinformatics tools to extract meaningful biological data. Among the many approaches in proteomics, targeted proteomics has greatly enhanced our understanding of systems biology and biomedical research, and its applications are increasingly being adopted in clinical settings.
Proteomic research often begins with bottom-up proteomics, a strategy where proteins are enzymatically digested into peptides for analysis. This approach forms the foundation for more targeted proteomic studies, where specific peptides or proteins are selectively quantified and analyzed. Bottom-up proteomics can involve label-free methods, which rely on intrinsic peptide properties for quantification, and stable isotope labeling techniques, which use isotopic tags to differentiate peptides and provide more accurate quantification.
Label-free quantification techniques offer a versatile approach to measure protein abundance without the need for chemical labeling, making them an attractive option for certain proteomic studies. These methods have evolved from semi-quantitative spectral counting, where protein levels are estimated based on the total number of assigned MS2 (tandem mass spectrometry) spectra, to a more accurate technique, extracted ion current in MS1 (first mass spectrometry) or MS2 scans. However, label-free proteomics has limitations, including variability between runs, challenges in detecting low-abundance proteins, and possible low reproducibility during sample preparation, separation, and data acquisition. Additionally, comprehensive data analysis can be complex and o en requires advanced bioinformatics tools for proper annotation.
Relative proteomic quantification using isobaric labeling technology has emerged as a preferred sample preparation method for processing complex biological matrices due to the limitations of label-free methods. Among the isobaric labeling technologies, Tandem Mass Tags stand out by facilitating sample multiplexing for Mass Spectrometry (MS)-based quantification and identification of biopolymers such as proteins, peptides, and nucleic acids.
Tandem Mass Tag (TMT) labeling developed by ThermoFisher Scientific is a well-established labeling reagent. The TMT reagent is designed for multiplexed quantitative proteomics, facilitating the simultaneous analysis of multiple peptide samples. It has a distinct structure with a mass reporter, cleavable linker, mass normalization segment, and a reactive group that specifically binds to the amino (-NH2) group of peptides for targeted labeling in mass spectrometry (Figure 1). These molecules have the same nominal mass but generate distinct reporter ions with different masses upon fragmentation. The relative proportions of these measured reporter ions correspond to the relative abundance of the labeled molecules, enabling precise quantification and comparison across multiple samples.
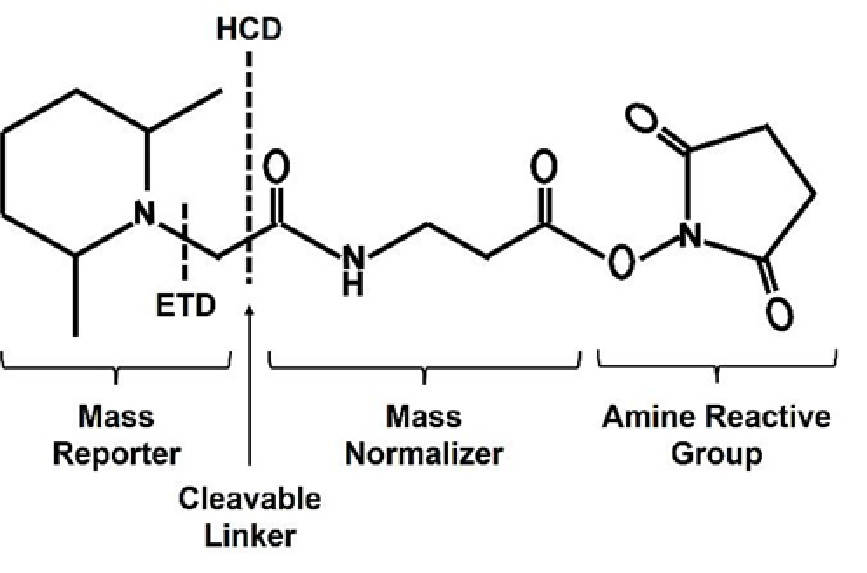
Figure 1: General chemical structure formula of the Tandem Mass Tag or TMT reagent (Source:
https://assets.thermofisher.com/TFSAssets%2FLSG%2Fmanuals%2FMAN0018773_TMTproMassTag
LabelingReagentsandKits_UG.pdf).
In TMT labeling, peptides derived from varied sources are labeled by linking di erent isotope-coded tags to either the N-terminus or the lysine residues of the peptides, typically at pH 8.5. This strategic labeling approach utilizes a combination of stable isotopes, including C13 and N15, ensuring a constant total molecular weight despite variations in isotopic composition.
Although all TMT tags have the same chemical structure, the isotopic substitutions create di erences in the molecular weights of the mass reporter and mass normalization regions. This allows labeled peptides to remain indistinguishable during chromatographic separation and single mass spectrometry (MS) analysis. In MS/MS mode, fragmentation yields both sequence information from the peptide backbone and quantification data from the mass reporter ions, facilitating accurate assessment of peptide abundances and robust comparative analyses.
Incorporating stable isotopes result in unique monoisotopic mass di erences of 6 mDa in the resulting reporter ions a er MS analysis. These subtle mass shi s are critical for accurate quantification and enable the di erentiation of peptide sources, even in complex biological samples. High-resolution mass spectrometry (HRMS) further enhances the precise measurement of these mass di erences, facilitating reliable quantification of relative abundances of peptides from different sources.
This multiplexing capability significantly improves throughput, enabling the analysis of multiple experimental conditions in a single run. By increasing e iciency and enabling deeper comparative analyses, TMT technology advances our understanding of biological processes and protein dynamics in proteomics research.
The TMT labeling begins with the preparation of protein samples derived from cell lines (treated and controls). A er extracting proteins and quantifying their concentration, the proteins are reduced and alkylated to facilitate their digestion. The proteins are then digested using an MS-compatible enzyme, such as trypsin, which cleaves them into tryptic peptides. These peptides are desalting and reconstituted in an amine-free bu er. The labeling reaction is initiated by adding specific TMT reagents and quenching the reaction with hydroxylamine. After the reaction, which stabilizes the labeled peptides, the pooled samples are subsequently vacuum dried, and aliquots are stored -80° C until analyzed via tandem mass spectrometry (MS/MS). The TMT labeling experimental workflow is shown in Figure 2.

Figure 2: Tandem Mass Tag (TMT) labeling experimental workflow. (Source: Deng et al Front. Mol. Neurosci. 12:24. doi: 10.3389/fnmol.2019.00024).
However, the effectiveness of TMT labeling can be influenced by various factors such as pH, temperature, and buffer composition during the labeling process. To optimize labeling efficiency, it is crucial to address the following challenges.
Additionally, simplifying sample composition can improve the sensitivity and specificity of MS analysis. One e ective approach is peptide prefractionation prior to MS analysis. High-performance liquid chromatography (HPLC) is employed to separate peptides into smaller fractions based on their physicochemical properties, such as hydrophobicity, charge, or size (Figure 3). This reduction in sample complexity minimizes competition for ionization during MS analysis, increasing the coverage of complex proteomes and improving the detection of low abundance peptides.
With the availability of commercial TMT tags that allow for the analysis of up to 18 protein samples simultaneously, it is imperative to implement a controlled workflow that includes refined sample preparation and accurate labeling procedures to ensure reliable and reproducible results.
Figure 3: High-performance liquid chromatography (HPLC) based fractionation of Tandem Mass Tag (TMT) labeled peptides.
Once labeled, the samples are analyzed using MS/MS analysis. In MS1 spectrum, peptides labeled with di erent TMT reagents appear as a single peak, facilitating simultaneous analysis across different samples. In the subsequent MS2 analysis, the chemical bonds between the TMT reporter group, balance group, and reaction group are cleaved, releasing the TMT reporter group and balance group, which enables the detection of reporter ions.
During data acquisition, the neutral loss of the balance group aids in detecting TMT reporter ions in the low-mass region of the MS2 spectrum. The intensity of these reporter ion peaks reflects the relative abundance of the peptides across di erent samples. Additionally, the mass-to-charge ratio of the peptide fragment ion peak in MS2 provides crucial sequence information about the peptides. Ultimately, qualitative, and relative quantitative information about the proteins can be obtained from these raw data through database retrieval, facilitating a comprehensive analysis of protein expression across samples. This structured approach ensures reliable results in proteomics research.
At Aragen, we specialize in high-quality proteomics sample preparation for quantitative analysis, utilizing both label-free and label-based methodologies. Our team of experienced proteomics scientists is skilled in handling a diverse range of specimen types, including cultured cells, tissues, organelles, and biofluids. We focus on optimizing peptide labeling conditions, minimizing sample loss, and ensuring accurate quantification, addressing the complexities inherent in proteomic analyses.
To ensure consistent and reliable results, we employ a robust quality control system that is integrated throughout our processes. Our state-of-the-art proteomics sample preparation facility is equipped with advanced technologies, including a high-performance liquid chromatography system with a fraction manager. This ensures the e icient separation and collection of peptides, meeting the stringent quality control standards required for accurate quantitative proteomics analysis.