
Covalent inhibitors are compounds designed to form a covalent bond with a specific molecular target. Covalent drugs (small molecule as well as protein/peptide) exhibit pharmacological advantages, such as prolonged duration of action and enhanced potency over non-covalent drugs, giving them a higher probability of efficacy in medicating hard-to-treat human diseases.
Despite the success of the earliest covalent drugs (e.g., aspirin, penicillin, and omeprazole — the first proton pump inhibitor), the pharmaceutical industry remained reluctant to develop covalent inhibitors for nearly 100 years. However, as technology and methodologies have evolved, the trend has reversed: to date, more than 100 covalent drugs have reached the market or progressed to advanced clinical trials (Fig. 1).
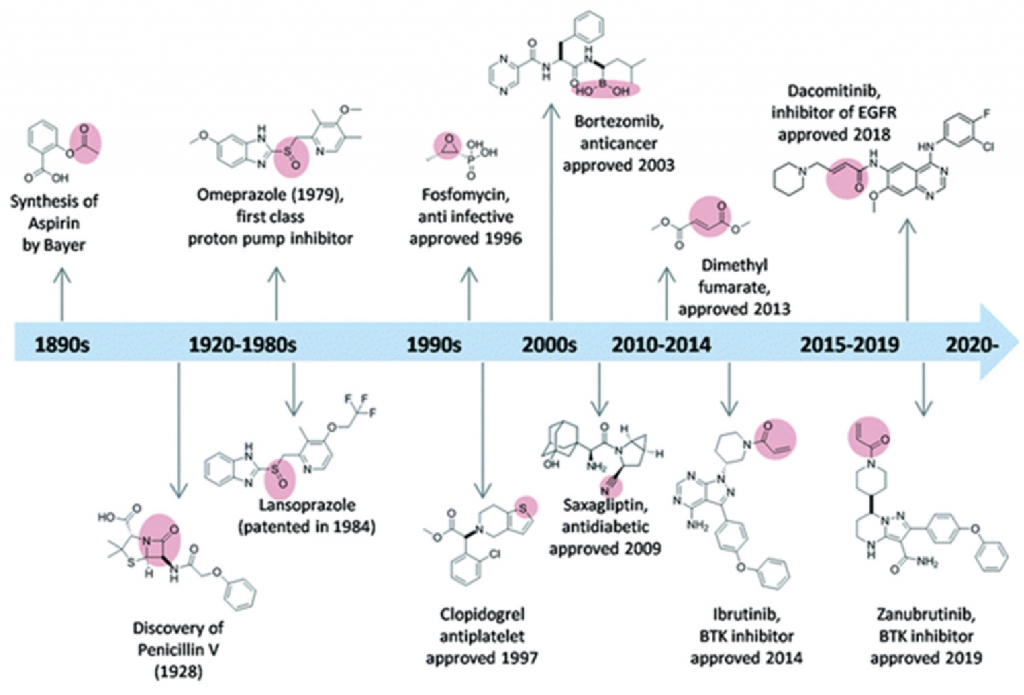
Moreover, because covalent therapies can be notoriously difficult to work with, most exploration and development in this area has been achieved by large pharma. By contrast, very little has been accomplished through contractual integrated drug discovery with a CRO. However, that dynamic is changing.
The methodologies and technology that drive covalent therapeutic development now are accessible to organizations of any size through partners like Aragen, which offer the knowledge base and the infrastructure to help those organizations succeed. By understanding covalent therapeutics’ inherent challenges and providing insight into how difficulties can be overcome, we empower our partners to seize opportunities in this rapidly expanding field.
With unique mechanistic properties that differentiate them from non-covalent inhibitors, covalent drugs can be administered at a lesser dose and frequency, reducing off-target events and increasing patient compliance. Covalent drugs also can be effective for low-vulnerability targets, wherein high levels of occupancy are needed to generate the desired physiological outcome, as well as effective in drugging chemically intractable targets. Additionally, covalent therapeutics can be a strategy to overcome drug-resistance mutations in gene-encoding the target protein (as in thecase of EFGR).
Covalent therapeutics have been most prominent, to date, in oncology, antivirals, and anti-infectives — areas where plenty of opportunity continues to exist. For example, a novel approach combining proteolysis targeting chimeras (PROTACs)[ii] and covalent drugs could provide advantages over non-covalent or PROTAC-only strategies. The recent approvals of Mirati KRASG12C and LUMYKRAS® (sotorasib) represent additional exciting applications of covalent therapeutics.[iii],[iv]
A key challenge associated with covalent therapeutics is toxicity.[v] Following a toxic event with a strong covalent drug, wash is very difficult. So, for days after the drug is no longer detected in the circulation, it still can be bound to the target and may elicit adverse events (AEs).
On one hand, covalent protein drug benefits can be difficult to demonstrate, partly because canonical amino acids present in peptide and protein drugs limit their chemical reactivity with target molecules. Accordingly, for many years, covalent therapeutics essentially arose out of serendipity — accidental findings, for example, for orphan disease states or diseases that did not have many clinical options.
On the other hand, a more prominent challenge is that electrophilic warheads[vi] — introduced to derive covalent fragments from their noncovalent predecessors and promote a reversible or irreversible covalent bond — can react with nucleophilic residues on proteins other than the intended target (e.g., conjugating with GSH), which can lead to off-target effects. In turn, these can lead to idiosyncratic toxicity and haptenization (immunogenic response).
A “hot” electrophilic warhead can potentially bind to every single nucleophilic residue on the protein that could result in reactive metabolites, which elicit toxicity and immune system response. Formerly, developers lacked the understanding to fine-tune that electrophilicity of the warhead. For example, a developer might have five basic amino acid pairs on a protein but only want to bind to one, leaving the others intact, even though they all are nucleophilic in nature.
While other biology challenges can arise — including concerns about stability, integrity (in vivo) administration routes, MOA (in vivo), PK-PD, ADME, and safety profiles in relevant disease models before IND qualification — modern methodologies and technology have positioned deliberately crafted, function-specific covalent therapies more consistently within reach. Additionally, the technology/instrumentation is more accessible now than ever before, and the parade of covalent therapies progressing through clinical trials and regulatory approval ensures precedent is available when seeking a regulatory pathway to market.
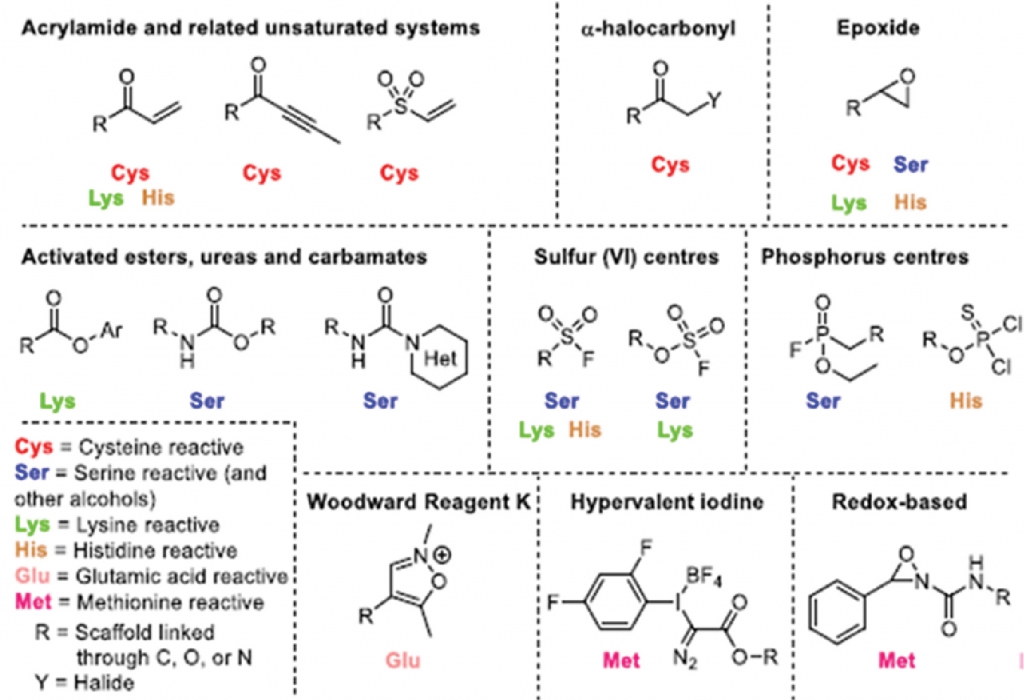
In recent years, the discovery of covalent drugs has been boosted by rational design and facilitated by the emergence of new technologies in proteomics, which enables identification of covalent drugs with higher selectivity by identifying their binding site. Traditionally, it believed that the warhead always had to attack the cysteine. Now, a multitude of warheads can be applied to target specific amino acid residues, enabling researchers to dial in the warhead’s electrophilicity and target other bases: lysine, glutamine, etc. — greatly expanding the opportunity to design better covalent compounds (Fig. 2).
In a new approach in the covalent therapeutics area, proximity-enabled reactive therapeutics (PERx) could provide ageneral platform technology for converting various interacting proteins into covalent binders, achieving specific covalent protein targeting for biological studies and enabling a new generation of covalent protein drugs. Li and coworkers showed that, through genetic code expansion, a latent bioreactive unnatural amino acid, fluorosulfate-L-tyrosine (FSY) can be incorporated into the ectodomain of the human programmed cell death protein-1 (PD-1). The resulting PD-1(FSY) covalently bound only to its natural ligand PD-L1 selectively via the reaction of its Histidine residue in a sulfur-fluoride exchange. A superior anti-tumor effect of the covalent PD-1(FSY) was seen both in vitro and in vivo compared to anti-PD1 antibody treatment. Control experiments showed that mutated PD-L1 (H69A), the covalent binding was absent indicating a high degree of residue specificity.[viii] Covalent inhibitors might target enzymes such as protein kinases, RAS proteins involved in human diseases, as potential treatment. Many programs consider pursuing that modality as a way to pin down a target that has been difficult to affix to a binder or inhibitor, because the affinity and the selectivity have not been there.
Covalent therapeutics offer the flexibility to attempt a low pill burden — a low amount or a smaller concentration. Once researchers elicit the pharmacology, it can have a perpetual effect in a positive way over time without having to have significant drugs on board. These therapeutics also can be realized as reversible covalents (noted above) or semi-covalents, a feature that helps tailor the approach for the disease at hand, maintaining efficacy without eliciting numerous AEs, compared to the past.
Current development of a variety of covalent inhibitors to address human health conditions, combined with recent FDA approval of several covalent therapeutics for use in humans, has energized interest in covalent therapeutics.
Aragen offers clients of all sizes the required components to build a successful program from design to implementation, through measurement of efficacy and liability, all the way into data analysis, to generate a compound with as high a probability as possible of becoming a potential drug.
The challenges associated with covalent therapeutics have not disappeared, but we have a deeper understanding of how to address those challenges, born of varied experience. We can significantly de-risk this strategy because of that experience — not only in the design, but also in the bioanalytical methods used to work with in this field, which are not unremarkable (e.g., measuring a compound containing boron is very challenging).
Even if a researcher can look for exposure of those compounds or their interaction with the target tissue, the refined bioanalytical methods to do so need to be developed. Furthermore, those methods should be devised by analytical scientists with experience in this field, which is unique from the more typical small-molecule inhibitor or small-molecule agonist programs.
When a pharma/biotech is attracted to covalent therapeutics, they need more than a partner that claims to understand covalency and how to design for it. They require a partner capable of developing a project that holds up downstream: measuring animal efficacy, measuring exposure in the tissues & circulation, examining AEs, predicting potential liabilities, etc. To learn more, contact the author and visit us at www.aragen.com.
Simon Haydar is Senior VP and Head of Integrated Drug Discovery Solutions at Aragen, to which he brings more than two decades’ experience in leading integrated drug discovery projects. Over the years, he has spearheaded discovery projects to develop novel oral therapies across a variety of therapy areas. Simon’s functional expertise spans areas including discovery R&D leadership, licensing due diligence, biotech alliances & partnerships, external R&D activities, and mergers & acquisitions. Prior to joining Aragen, Simon held leadership roles at Syngene International Limited, Assembly Biosciences, Eli Lily, GSK, and Pfizer.
Aragen is a global leader in providing drug discovery, drug development, and manufacturing solutions for life sciences firms.